
")
")


Virtual screening and rational design of protein-protein interaction modulators
with balanced ADME-Tox properties
Team 3 led by Dr. M. Miteva (DR Inserm)
Team's publications / Organization Chart
The search for innovative therapeutic molecules and pharmacological probes that help to understand better the role of a target in a pathophysiological mechanism is definitively required considering that only about a quarter of the 30,000 listed diseases in the world has an appropriate treatment. Indeed, several approaches can be used to identify and optimize drug candidates rationally.
In our team, to meet some of these challenges and the associated scientific issues, we have chosen to combine in silico screening and multiparameter optimization protocol with pre-clinical and clinical approaches. Many applications on innovative therapeutic targets are being studied with our experimentalists partners in France and in the world.
We apply in the team a disciplinary and conceptual continuum to several therapeutic areas, mainly cancer, cardiovascular (coagulation) disease and certain rare diseases. We carry methodological developments in silico associated with our scientific questions and we focus our work on innovative molecular mechanisms: the modulation of protein-protein interactions with small molecules. This category of targets offers unique opportunities with approximately 500,000 protein-protein interactions critical for human life and the need to rationally explore new areas of chemical space comprising several billion compounds. The goal is to be able to develop some small molecules up to early stage clinical trials.
To date, our partners or internally in our team, we have already produced promising results, both about new in silico approaches and on novel targets. We have several molecules in various stages of development, from the stages of in silico-in vitro studies to experimental evaluation using animal models on about 10 therapeutic targets. Our work focuses on the methodological development and applications in human health, as evidenced by our patents and publications.
The issues we address in our team to further contribute to therapeutic innovation and human health are highly complementary:
Theme 1 : Exploring the chemical space and virtual screening for the modulation of the protein-protein interactions (PPis).
Theme 2 : Rational development and optimization of therapeutic molecules under ADME-Tox constraints and considerations of genetic variability (pharmacogenomics, pharmacogenetics).
Theme 3 : Using the knowledge generated in themes 1 and 2 for the identification of innovative bioactive molecules in 3 therapeutic areas: cancer, cardiovascular diseases and rare diseases.
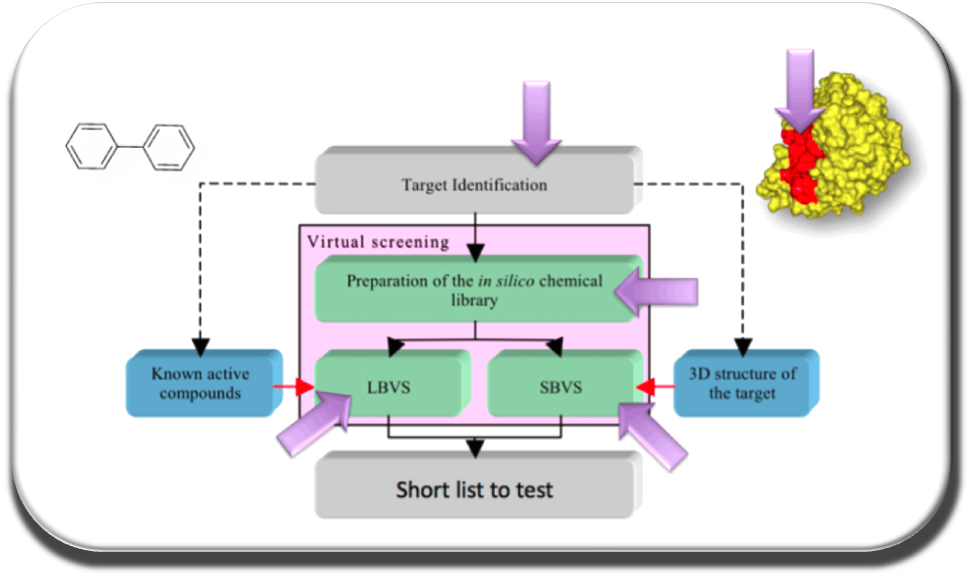
Our team focus on :
In the continuity of our previous work, we develop software tools for drug design in optics to target PPIs with appropriate ADME-Tox properties. Recently we developed the first free webserver in France dedicated to screen in silico a large number of drug-like or “PPI-friendly” compounds, MTiOpenScreen (Labbe et al. NAR 2015) via the RPBS platform (in collaboration with team 1 of MTi). To efficiently target flat and flexible PPIs surfaces we elaborate original protocols combining molecular dynamics, free energy calculations and druggability prediction to identify new druggable pockets at the PPI interfaces (Zhang et al. Plos One 2014). We have established a new protocol to consider protein flexibility using molecular dynamics simulations and normal mode analysis depending on local or global conformational changes that can occur due to ligand binding (Moroy et al. Future Med Chem 2015). Our new version of AMMOS v. 2 makes it possible to improve the prediction of protein-ligand interactions taking into account local protein flexibility and water molecules key for the interaction (Pencheva et al, Curr Comput Aided Drug Design, 2013; Jereva et al. As J Phys 2014) (in collaboration with Dr.Pencheva and Pr. Pajeva, Bulgarian Acad. Sci).
In order to develop new in silico drug design tools, it is important to be aware of existing approaches. We have collected and evaluated for over 10 years over 1700 in silico tools that assist drug discovery. The study has been reported in Drug Discovery Today 2013, Villoutreix et al. This list of about 1700 free in silico tools and databases is available to the scientific community on the VLS3D website.
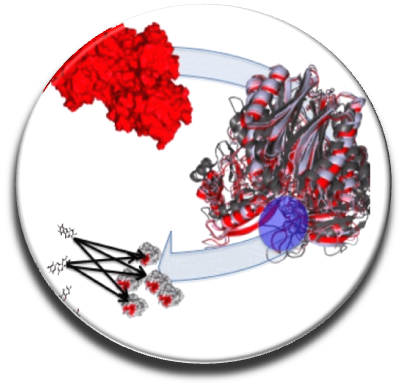
Low molecular weight «drug-like» modulators of protein-protein interactions (PPIs)
ADME-Tox Prediction
 The quality of compound collections is essential to increase the chances of discovering potent drug candidates or to find probes. The underlying concept is: if molecules of bad qualities are used for screening (toxic, reactive molecules, frequent hitters…) without being aware that they contain such groups, the hits found may not be very relevant to develop a drug or a pharmacological probe. To facilitate this process, we developed a new version of the first online server FAF-Drugs, v.3 (Lagorce et al., NAR 2015) that filters out unwanted small molecules by calculating the physicochemical properties and searching for structural alerts (toxic/reactive groups, « frequent hitters »), or for groups interfering with the experimental screening tests (e.g., PAINS compounds in collaboration with Pr. Baell, Monash University, Australia) (there are other servers but in general they can only process 1 compound at a time, eventually up to 1000 molecules or they output only a small number of parameters like logP).
The quality of compound collections is essential to increase the chances of discovering potent drug candidates or to find probes. The underlying concept is: if molecules of bad qualities are used for screening (toxic, reactive molecules, frequent hitters…) without being aware that they contain such groups, the hits found may not be very relevant to develop a drug or a pharmacological probe. To facilitate this process, we developed a new version of the first online server FAF-Drugs, v.3 (Lagorce et al., NAR 2015) that filters out unwanted small molecules by calculating the physicochemical properties and searching for structural alerts (toxic/reactive groups, « frequent hitters »), or for groups interfering with the experimental screening tests (e.g., PAINS compounds in collaboration with Pr. Baell, Monash University, Australia) (there are other servers but in general they can only process 1 compound at a time, eventually up to 1000 molecules or they output only a small number of parameters like logP).
Therapeutic targets
- Cancer
We identified two molecules, which inhibit the activity of Bfl-1 and stimulate the death of cancer cells (Mathieu et al, J Biomol Screen. 2014) (in collaboration with Pr. Deprez, Inserm U761, Lille, and Dr. Bonnefoy, Inserm U503, Lyon). In collaboration with Dr. Colas (CNRS, Roscoff) we discovered two new molecules blocking the CDK-CKS protein interaction and the proliferation of tumor cells (Hamdi et al. Chembiochem 2015). In collaboration with Dr. Starzec and Pr. Perret (Univ. Paris 13) we identified anti-angiogenic molecules which inhibit the VEGF-neuropiline interaction (Starzec et al. Bioorg Med Chem 2014). We recently reported novel anti-cancer molecules (Lindstrom et al. Mol. Cancer Res. 2015) (in collaboration with Dr Alvarado-Kristensson (University Hospital, Malmö) and repositioning of a FDA approved drug for anti-cancer application.
- Design of novel pro and anticoagulant chemical probes through in silico and in vitro screening
Blood coagulation involves a series of reactions between enzymes and cofactors assembled on certain cell surfaces . Coagulation is activated by different molecular mechanisms and results in the generation of locally high concentration of thrombin, the enzyme that coagulates the blood in part by conversion of soluble fibrinogen to insoluble fibrin. Several anticoagulant control mechanisms are operating and under normal conditions , the systems are in equilibrium : coagulation and bleeding are avoided (Villoutreix & Dahlback , Arteriosclerosis, Thrombosis , and Vascular Biology 2005). Hereditary and acquired conditions can tip the balance in favor of anticoagulant mechanisms causing bleeding or procoagulant mechanisms and the associated risk of thrombosis (about 20,000 people die of DVT each year in France). The principle used for the therapeutic treatment of coagulation disorders such as hemophilia is injecting the missing clotting factor, while the inhibition of coagulation factors is the dominant approach for the treatment of thrombosis.
- Rare diseases
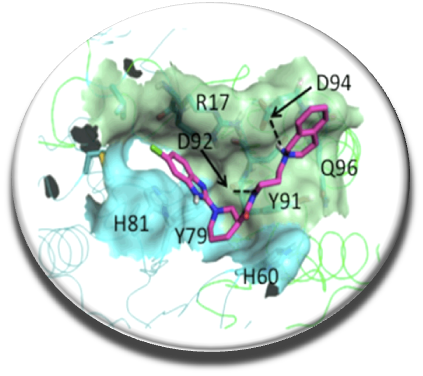
Recently we reported the first identified molecules which play a role of stabilizers of the dimer of spermine synthase (SMS) (Zhang et al. Plos One 2014). It was discovered that some mutations of SMS are responsible for a rare mental disease, the Snyder-Robinson syndrome. In collaboration with Pr. Alexov (Clemson Univ., USA) and Dr. Ikeguchi (Josai Univ., Japan) we demonstrated that these molecules increase the protein activity via the dimer stabilization. Predicted binding mode of the compound E941-0318 increasing the activity of the destabilized SMS G56S dimer.
- Design of anticoagulant probes : inhibition of protein-membrane interactions
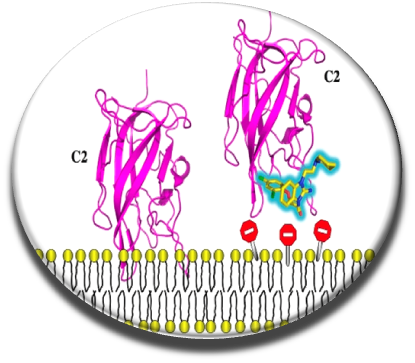
Effective anticoagulant drugs have been developped these last 50 years (heparin, vitamin K antagonists or more recently direct inhibitors of thrombin and factor X) , but they often exhibit undesirable side effects and can not be administered to all patients. Rather than carry out work on the conventional therapeutic targets and attempt to inhibit the catalytic sites of serine proteases (Villoutreix & Sperandio, Curr Opin Struct Biol. 2010), we focused on our team and in collaboration with Dr. G. Nicolaes from the Cardiovascular Institute of the University of Maastricht on innovative mechanisms, including inhibition of interactions between clotting cofactors V and VIII and procoagulant cell membranes. This interaction is crucial for the generation of thrombin and we thought that impeding this contact with a small molecule could be of interest. The key protein domain for interaction with the membranes of factor V and factor VIII is the C2 domain. We screened in silico this domain (docking - scoring and post-processing) using a compound collection of hundreds of thousands of compounds and tested in vitro a few hundred molecules (Segers et al . PNAS 2007). About 10 small anticoagulant chemical molecules were identified, these compounds were effective in vitro but did not work on whole blood, partly due to a strong interaction with a plasma protein.
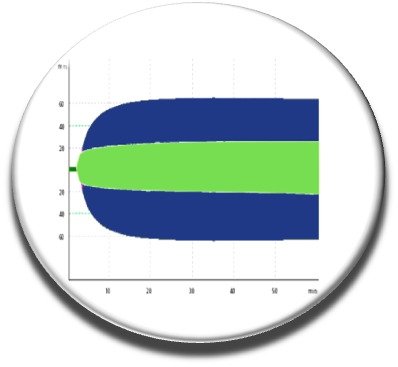
We continued our work by coupling several chemoinformatic approaches (pharmacophore, fingerprint, docking), in vitro screening, experimental and biophysical approaches to arrive in 2014 with new molecules that inhibit the interaction of factor V and factor VIII with membranes (Nicolaes et al, Blood 2014). The C11-10 molecule seems to work on whole blood as seen by thromboelastography experiments carried out on the blood of healthy individuals without compound C11-10 (blue curve) or in the presence of this small chemical probe (green curve). We have a proof of concept that need to be explored further and should help to design novel anticoagulant molecules.
- Design of procoagulant chemical probes: inhibition of protein-protein interactions
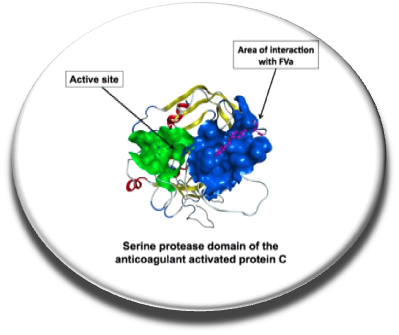 In cases of inherited or acquired bleeding, it would be interesting to have small orally available small molecules to reduce the number of injections of certain proteins (eg, factor VIII ) or to replace the administration of therapeutic proteins when the patient can not tolerate this type of therapy. The protein C anticoagulant system is an essential mechanism, indeed activated protein C form with protein S an anticoagulant complex that inactives by factors Va and VIIIa and thereby inhibiting the generation of thrombin. Protein C deficient patients are particularly prone to thrombosis. In fact inhibiting the active site of protein C increases the in vitro thrombin generation in hemophiliac however inhibiting the catalytic site of this serine protease damage the cytoprotective fonction of protein C. We screened in silico and in vitro a protein C exosite important for the interaction with factor Va and identified procoagulant small molecules blocking this protein-protein interaction. The binding site of the compounds was studied by SPR in the presence of wild-type protein C, protein C with mutations in the exosite area and the bioactive compounds. This proof of concept is encouraging and suggests that hemophilia treatment with orally available molecules could be developed and that such small molecules could also be useful as an antidote to some anticoagulant drugs (Sperandio et al., Thrombosis Research, 2014).
In cases of inherited or acquired bleeding, it would be interesting to have small orally available small molecules to reduce the number of injections of certain proteins (eg, factor VIII ) or to replace the administration of therapeutic proteins when the patient can not tolerate this type of therapy. The protein C anticoagulant system is an essential mechanism, indeed activated protein C form with protein S an anticoagulant complex that inactives by factors Va and VIIIa and thereby inhibiting the generation of thrombin. Protein C deficient patients are particularly prone to thrombosis. In fact inhibiting the active site of protein C increases the in vitro thrombin generation in hemophiliac however inhibiting the catalytic site of this serine protease damage the cytoprotective fonction of protein C. We screened in silico and in vitro a protein C exosite important for the interaction with factor Va and identified procoagulant small molecules blocking this protein-protein interaction. The binding site of the compounds was studied by SPR in the presence of wild-type protein C, protein C with mutations in the exosite area and the bioactive compounds. This proof of concept is encouraging and suggests that hemophilia treatment with orally available molecules could be developed and that such small molecules could also be useful as an antidote to some anticoagulant drugs (Sperandio et al., Thrombosis Research, 2014).
