
")
")


Virtual screening and rational design of protein-protein interaction modulators
with balanced ADME-Tox properties
Equipe 3 du Dr. M. Miteva (DR Inserm)
Publications de l'équipe / Organigramme
La recherche de molécules thérapeutiques innovantes et de sondes pharmacologiques permettant de mieux appréhender le rôle d’une cible dans un mécanisme pathophysiologique est une nécessité absolue en considérant qu’environ seulement un quart des 30000 maladies répertoriées dans le monde dispose d’un traitement pertinent. Plusieurs approches doivent être utilisées pour identifier des candidats médicaments et les optimiser de manière rationnelle.
Dans notre équipe, pour répondre à certains de ces défis et aux questions scientifiques associées, nous avons fait le choix de coupler les approches in silico de type criblage virtuel et l’optimisation multiparamétrique avec une perspective d'ouverture expérimentale pré-clinique et clinique. De nombreuses applications sur des cibles thérapeutiques innovantes sont actuellement étudiées avec nos partenaires expérimentalistes en France et dans le monde.
Nous appliquons ainsi dans l’équipe un continuum disciplinaire et conceptuel dans plusieurs domaines thérapeutiques, essentiellement le cancer, maladies cardiovasculaires et certaines maladies rares. Nous réalisons des développements méthodologiques in silico associés à nos questions scientifiques et nous centrons nos travaux sur des mécanismes moléculaires novateurs de type modulation des interactions protéine-protéine par des petites molécules chimiques. Cette catégorie de cibles offre des opportunités uniques avec environ 500,000 interactions protéine-protéine critiques pour la vie humaine et le besoin d’explorer rationnellement de nouvelles zones d’un espace chimique comprenant plusieurs milliards de composés. L’objectif est de pouvoir développer certaines petites molécules jusqu’au stade des essais thérapeutiques précoces. A ce jour, avec nos partenaires ou en interne dans notre équipe, nous avons déjà des résultats prometteurs, aussi bien sur les approches silico que sur les applications sur cibles. Nous disposons ainsi de plusieurs molécules à différents stades de développement, depuis le stade in silico, in vitro jusqu’à l’évaluation expérimentale à l’aide de modèles animaux et ceci sur plus de 10 cibles thérapeutiques. Nos travaux sont orientés sur le développement méthodologique avec des applications dans la santé humaine, comme l’atteste nos brevets ainsi que nos publications.
Les thèmes que nous abordons pour contribuer plus encore à l’innovation thérapeutique et à la Santé humaine sont éminemment complémentaires :
Thème 1 : Exploration de l’espace chimique et criblage virtuel pour la modulation des interactions protéine-protéine.
Thème 2 : Développement rationnel et optimisation de molécules thérapeutiques sous contraintes ADME-Tox & variabilité génétique (pharmacogenetics,pharmacogemonics).
Thème 3 : Utilisation des connaissances générées dans les thèmes 1 et 2 pour la recherche et l’évaluation de molécules bioactives innovantes dans 3 aires thérapeutiques fédératrices que sont le cancer, les maladies cardio-vasculaires et les maladies rares.
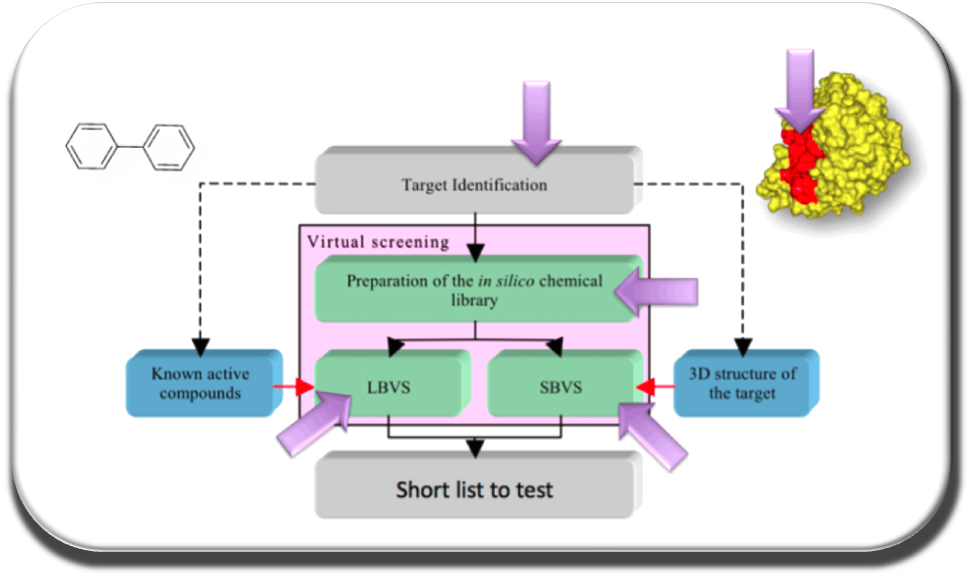
Notre équipe travaille en priorité sur :
Développement et optimisation des outils in silico pour le criblage virtuel afin de cibler les IPPs
Dans la continuité de nos travaux précédents, nous avons développé le premier serveur web gratuit en France dédié à cribler in silico un grand nombre de composés « drug-like » ou «PPI-friendly", MTiOpenScreen (Labbe et al. NAR 2015), disponible sur la plate-forme RPBS (en collaboration avec l'équipe 1 de MTi). Pour cibler efficacement les IPPs ayant des interfaces plats et flexibles nous élaborons des protocoles originaux combinant la dynamique moléculaire, calculs d'énergie libre et la prédiction de druggabilité afin d'identifier de nouvelles poches druggables aux interfaces PPIs (Zhang et al. Plos One 2014). Nous avons établi un nouveau protocole de considérer la flexibilité des protéines en utilisant des simulations de dynamique moléculaire et de l'analyse de modes normaux en fonction des changements conformationnels locaux ou globaux qui peuvent se produire lors de l’interaction avec un ligand (Moroy et al. Future Med Chem 2015). Notre nouvelle version de AMMOS v 2 permet d'améliorer la prédiction des interactions protéine-ligand en tenant compte de flexibilité de protéines et molécules d'eau clés pour l'interaction (Pencheva et al, Curr Comput Aided Drug Design, 2013; Jereva et al. As J Phys 2014) (en collaboration avec le Dr. Pencheva et le Pr. Pajeva, Acad. Bulgare des Sciences).
Afin de développer de nouvelles approches in silico pour le drug design, il est important de pouvoir analyser les approches existantes. Nous avons collecté et évalué depuis 10 ans plus de 1700 outils in silico qui aident la découverte de médicaments. Cette revue a été publiée dans Drug Discovery Today 2013, Villoutreix et al. Une liste d'environ 1700 outils in silico et bases de données est disponible à la communauté scientifique sur le site VLS3D.
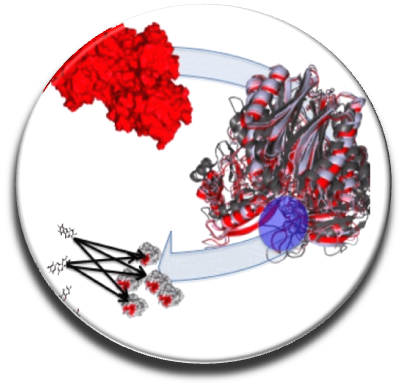
Petites molécules «drug-like» modulateurs d’intéractions protéines-protéines (IPPs)
Prédiction ADME-Tox
 Pour pouvoir sélectionner des molécules avec un profil «drug-like» pour le criblage (i.e. biodisponibles et pas toxiques), nous avons développé une nouvelle version du premier serveur en ligne FAF-Drugs3 (Lagorce et al., NAR 2015) qui implique des règles physico-chimiques simples inspirées des règles de Lipinski et comprend de plus un jeu d’environ 300 règles pour detecter des composés qui comprennent des groupes toxiques/réactifs, « frequent hitters », ou PAINS (en collaboration avec le Pr. Baell, Monash University, Australie) . FAF-Drugs3 est le premier outil en ligne qui permet le filtrage d’un grand nombre de composés (d’autres outils existent mais ne traitent qu’un petit nombre de molécules et/ou ne calculent que peu de paramètres/descripteurs comme par ex. logP).
Pour pouvoir sélectionner des molécules avec un profil «drug-like» pour le criblage (i.e. biodisponibles et pas toxiques), nous avons développé une nouvelle version du premier serveur en ligne FAF-Drugs3 (Lagorce et al., NAR 2015) qui implique des règles physico-chimiques simples inspirées des règles de Lipinski et comprend de plus un jeu d’environ 300 règles pour detecter des composés qui comprennent des groupes toxiques/réactifs, « frequent hitters », ou PAINS (en collaboration avec le Pr. Baell, Monash University, Australie) . FAF-Drugs3 est le premier outil en ligne qui permet le filtrage d’un grand nombre de composés (d’autres outils existent mais ne traitent qu’un petit nombre de molécules et/ou ne calculent que peu de paramètres/descripteurs comme par ex. logP).
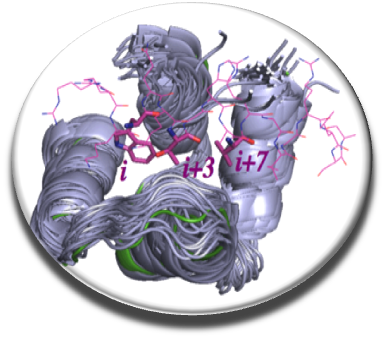
Cibles thérapeutiques
- Cancer
Nous avons identifié deux molécules qui inhibent l'activité de Bfl-1 et stimulent la mort des cellules cancéreuses (Mathieu et al, J Biomol Screen. 2014) (en collaboration avec le Pr. Deprez, Inserm U761, Lille, et le Dr Bonnefoy, Inserm U503, Lyon). En collaboration avec le Dr. Colas (CNRS, Roscoff), nous avons découvert deux nouvelles molécules bloquant l’interaction protéique CDK-CKS et la prolifération des cellules tumorales (Hamdi et al. Chembiochem 2015). En collaboration avec le Dr Starzec et le Pr. Perret (Univ. Paris 13), nous avons identifié des molécules anti-angiogéniques qui inhibent l'interaction VEGF-neuropiline (Starzec et al. Bioorg Med Chem 2014). Nous avons récemment rapporté molécules anticancéreuses (Lindstrom et al. Mol. Cancer Res. 2015) (en collaboration avec le Dr Alvarado-Kristensson (University Hospital, Malmö) et le repositionnement d'un médicament approuvé par FDA pour une application anti-cancer .
- Design de sondes chimiques anti- et pro-coagulantes innovantes par criblage in silico et in vitro
La coagulation sanguine comprend une série de réactions entre des enzymes et des cofacteurs assemblés sur certaines surfaces cellulaires. L’activation de la coagulation par différents mécanismes moléculaires entraîne la production de concentration localement élevée de thrombine, enzyme qui coagule le sang en partie par la conversion du fibrinogène soluble en fibrine insoluble. Plusieurs mécanismes anticoagulants contrôlent la coagulation et dans les conditions normales, les systèmes sont en équilibres : le saignement et la coagulation sont évités (Villoutreix & Dahlback, Arteriosclerosis, Thrombosis, and Vascular Biology 2005). Des maladies héréditaires et acquises peuvent faire pencher la balance en faveur de mécanismes anticoagulants entraînant une hémorragie ou de mécanismes procoagulants avec risques de thrombose (environ 20000 personnes décèdent de thrombose veineuse chaque année en France). Le principe thérapeutique utilisé pour le traitement des troubles de la coagulation comme l’hémophilie est d’injecter le facteur de coagulation manquant, alors que l’inhibition des facteurs de coagulation est l’approche dominante pour le traitement de la thrombose.
- Maladies rares
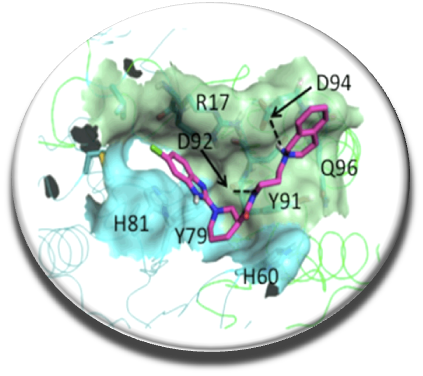
Les IPPs représentent une nouvelle classe de cibles émergentes insuffisamment exploitée pour des traitements thérapeutiques. Récemment, nous avons identifiées les premières molécules qui jouent un rôle de stabilisateurs du dimère de spermine synthase (SMS) (Zhang et la. Plos One 2014). Il a été découvert que certaines mutations de SMS sont responsables d'une maladie mentale rare, le syndrome de Snyder-Robinson. En collaboration avec le Pr. Alexov (Clemson Univ., USA) et le Dr. Ikeguchi (Josai Univ., Japon), nous avons demontré que ces molécules augmentent l'activité de la protéine via une stabilisation du dimère.
- Design de sondes anticoagulantes : inhibition d’interactions proteine-membrane
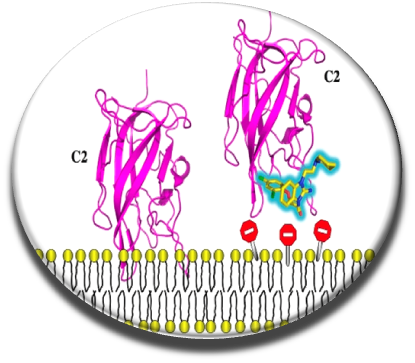
Des médicaments anticoagulants efficaces existent (l’héparine, les antagonistes de la vitamine K ou les inhibiteurs de la thrombine et du facteur X), mais ils présentent souvent des effets secondaires indésirables et ne peuvent pas etre administrés à tous les patients. Plutôt que de mener des travaux sur les cibles thérapeutiques classiques de type inhibition des sites catalytiques des serines protéases de la coagulation (Villoutreix & Sperandio, Curr Opin Struct Biol. 2010), nous nous sommes concentrés dans notre équipe et en collaboration avec le Dr. G. Nicolaes de l’Institut cardiovasculaire de l’université de Maastricht sur des mécanismes originaux, notamment l’inhibition des interactions entre les cofacteurs VIII et V de la coagulation et les membranes cellulaires procoagulantes. Cette interaction est cruciale pour la génération de thrombine et bloquer ce contact protéine-membrane nous semblait être une approche pertinente. Le domaine clef pour l’interaction avec les membranes du facteur V et du facteur VIII est le domaine C2. Nous avons criblé in silico le domaine C2 (docking-scoring et post-processing) en utilisant une chimiothèque de plusieurs centaines de milliers de composés et testé in vitro quelques centaines de molécules (Segers et al., PNAS 2007). Environ une dizaine de petites molécules chimiques anticoagulantes ont été identifiées, ces composés étaient efficaces in vitro mais ne fonctionnaient pas sur sang complet, en partie due à une interaction forte avec une protéine du plasma
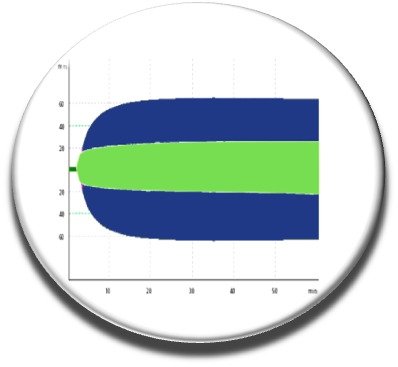
Nous avons continué nos travaux en couplant plusieurs approches chémoinformatiques (approches pharmacophores, fingerprint, docking), le criblage vitro, les approches expérimentales et biophysiques pour aboutir en 2014 à des nouvelles molécules inhibant l’interaction du facteur V et du facteur VIII avec les membranes (Nicolaes et al, Blood 2014). La meilleure molécule C11-10 marche sur sang complet comme le montre les résultats de thromboélastogrammes réalisés sur du sang d’individus sains sans la molécule C11-10 (courbe bleue) ou en présence de la petite sonde chimique (courbe verte). Nous espérons sur la base de ces travaux, développer des anticoagulants novateurs.
- Design de sondes procoagulantes : inhibition d’une interaction protéine-protéine
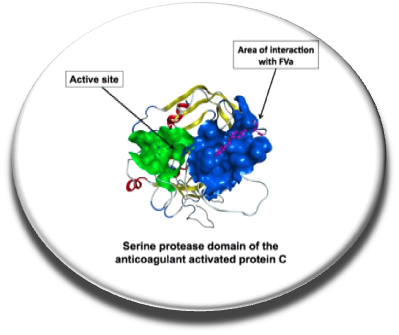 Dans les cas de saignements d’origine héréditaire ou acquise, il serait intéressant de disposer de petites molécules absorbables par voie orale pour par exemple réduire les injections de certaines protéines (e.g, le facteur VIII) ou pour remplacer l’administration de protéines thérapeutiques dans les cas ou le patient ne tolère pas ce type de thérapie. La voie de la protéine C est un système anticoagulant essentiel, en effet la protéine C activée forme avec la protéine S un complexe anticoagulant qui inactive par hydrolyse les facteurs Va et VIIIa et donc bloque la génération de thrombine. Les malades déficitaires en protéine C sont notamment sujets aux thromboses. En fait, bloquer le site actif de la protéine C augmente la génération de thrombine in vitro chez les hémophiles cependant en inhibant le site catalytique de cette serine protéase on inhibe aussi une autre fonction clef cytoprotectice de la protéine C. Nous avons criblé in silico et in vitro un exosite de la protéine C important pour l’interaction avec notamment le facteur Va et identifié des petites molécules procoagulantes bloquant cette interaction proteine-proteine. Le site de fixation des composés a été étudié par SPR en présence d’une protéine C normale, d’une protéine C mutée dans l’exosite et des composés bioactifs. Cette preuve de concept est encourageante et suggère que des traitements avec des molécules absorbables par voie orale pourraient être développés ou que ce type de petites molécules pourrait être intéressant comme antidote de certains médicaments anticoagulants (Sperandio et al., Thrombosis Research, 2014)..
Dans les cas de saignements d’origine héréditaire ou acquise, il serait intéressant de disposer de petites molécules absorbables par voie orale pour par exemple réduire les injections de certaines protéines (e.g, le facteur VIII) ou pour remplacer l’administration de protéines thérapeutiques dans les cas ou le patient ne tolère pas ce type de thérapie. La voie de la protéine C est un système anticoagulant essentiel, en effet la protéine C activée forme avec la protéine S un complexe anticoagulant qui inactive par hydrolyse les facteurs Va et VIIIa et donc bloque la génération de thrombine. Les malades déficitaires en protéine C sont notamment sujets aux thromboses. En fait, bloquer le site actif de la protéine C augmente la génération de thrombine in vitro chez les hémophiles cependant en inhibant le site catalytique de cette serine protéase on inhibe aussi une autre fonction clef cytoprotectice de la protéine C. Nous avons criblé in silico et in vitro un exosite de la protéine C important pour l’interaction avec notamment le facteur Va et identifié des petites molécules procoagulantes bloquant cette interaction proteine-proteine. Le site de fixation des composés a été étudié par SPR en présence d’une protéine C normale, d’une protéine C mutée dans l’exosite et des composés bioactifs. Cette preuve de concept est encourageante et suggère que des traitements avec des molécules absorbables par voie orale pourraient être développés ou que ce type de petites molécules pourrait être intéressant comme antidote de certains médicaments anticoagulants (Sperandio et al., Thrombosis Research, 2014)..
